 Photo credit to MLobliner via Flickr
Photo credit to MLobliner via Flickr
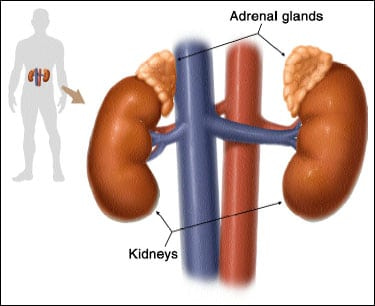
Pheochromocytomas are rare nervous system tumors that are usually benign and develop in the small glands that sit on top of the kidneys, called the adrenal glands. Everyone has two adrenal glands that produce hormones that travel throughout the body to give instructions to other organs and tissues. Tumors are often classified based on the body system they originate from. Pheos are considered neuroendocrine tumors, originating from nervous (neuro) and endocrine (hormone-producing). NeuroEndocrine Tumors are often abbreviated NET.  Photo credit to NET Cancer Day
Photo credit to NET Cancer Day
While pheos are usually benign (non-cancerous), it is possible for them to be cancerous. Most people who develop a pheo are between the ages of 20 and 50, but this tumor type can develop at any age.
Because pheos can cause the body to release too many “fight or flight” hormones, symptoms may include: high blood pressure, sweating, rapid heartbeat, tremors, shortness of breath and headache. These symptoms often occur in brief spells of 15 to 20 minutes. Spells can happen several times a day or less often.
Certain activities, situations, foods or medications may trigger symptoms, including:
- Physical exertion
- Anxiety or stress
- Changes in body position
- Bowel movements
- Labor and delivery
- Certain medications
- Certain foods that are contain a high amount of tyramine, a substance that is common in foods that are fermented, aged, pickled, cured, overripe or spoiled.
If left untreated, a pheo can result in severe or life-threatening damage to other body systems, especially the cardiovascular system. Surgery to remove the tumor is usually required.
Most individuals who develop a pheo do not have any previous history of these tumors or any family history of these tumors. However, research has shown that even in these apparently isolated cases, at least 1/4 individuals who develop pheos are born with a hereditary predisposition to develop these tumors. Red flags for a hereditary reason for pheo devleopment include younger age of diagnosis, multifocal tumors, and the presence of paraganglioma, another type of neuroendocrine tumor, in a personal or family with pheos. Learning about a possible hereditary predisposition is important for that individual’s medical care as well as that of their family members.
Pheos can be associated with the following hereditary syndromes:
1. von Hippel-Lindau disease (VHL)
- Caused by mutations within the VHL gene
- Average age of pheo diagnosis is 30 years
- Other clinical findings may include hemangioblastomas, which are benign tumors of the brain, spinal cord or eyes, as well as kidney cancer and tumors of the pancreas.
- Relatively high rate of new (de novo) VHL mutations, meaning that the mutation occurs for the first time in that individual. This may explain why features of this syndrome are often not seen in previous generations
2. Multiple Endocrine Neoplasia Type 2 (MEN2)
- Associated with mutations in the RET gene.
- There are 3 subtypes of MEN2- MEN2A, MEN2B, AND FMTC. Pheos can be see in MEN2A and MEN2B.
- Pheos commonly diagnosed between 30-40 years of age and can be the first sign in 9-27% of cases
- Other clinical findings may include medullary thyroid cancer and overactive parathyroids glands
3. Hereditary Paraganglioma-Pheochromocytoma (PGL-PCC) Syndrome
- Characterized by benign tumors in structures called paraganglia. Paraganglia are groups of cells that are found near nerve cell bunches called ganglia. A tumor involving the paraganglia is known as a paraganglioma. Like pheos, some paragangliomas cause overproduction of hormones.
- A Pheochromocytoma is a type of paraganglioma. A paraganglioma is called a pheo when it is found in the adrenal glands.
- This syndrome can be caused by mutations in many genes, including SDHB, SDHC, SDHD, SDHAF2, MAX, SDHA, and TMEM127.
- Tumors in this syndrome are typically diagnosed at earlier ages than when they develop by chance.
- Data are available on whether a mutation in each particular PGL-PCC gene is more likely to cause increased risk of paragangliomas versus pheos, tumors of the head and neck versus tumors of the abdomen and pelvis, tumors that causes overproduction of hormones versus those that usually do not, and tumors that are usually benign versus those that are more often malignant.
- If a mutation in either SDHD or SDHAF2 is inherited from one’s father, there is an increased risk of tumors. If a mutation in one of these genes is inherited from one’s mother, there is usually not an increased risk of these tumors. This is called a parent-of-origin effect.
{kind=link}
4. Neurofibromatosis 1
- Caused by mutations in the NF1 gene
- Characterized by multiple coffee-with-milk colored, flat skin findings, freckling in the armpits and groin area, multiple/usually benign tumors of nerves (neurofibromas). and pigmented nodules found on the iris of the eye called Lisch nodules.
- The presence of pheos in NF1 is much lower than other hereditary pheo syndromes
Some experts recommend that all people with pheos be offered genetic testing. Genetic testing is most likely to be positive if following charcateristics are present in person/family:
- Pheo diagnosis before age 50
- Multifocal disease
- Families with multiple cases of pheos and/or paranaglioma
- Personal and/or family history of any of the above additional clinical findings
- Known family history of any of the above syndromes
References:
JNCI (2003) 95 (16): 1196-1204
NEJM (2002) 346 (19): 1459-1466
J Hum Hypertens (2013) 27:141–147
Horm Metab Res (2012) 44:328–333
Clin Cancer Res (2012) 18:2828–2837
Clin Endocrinol (Oxf) (2013) 79(6):817-23
Nat Genet (2010) 42:229–233
Endocrinology (2011) 152:2133–2140
Curr Hypertens Rep (2010) 12(6)456-464